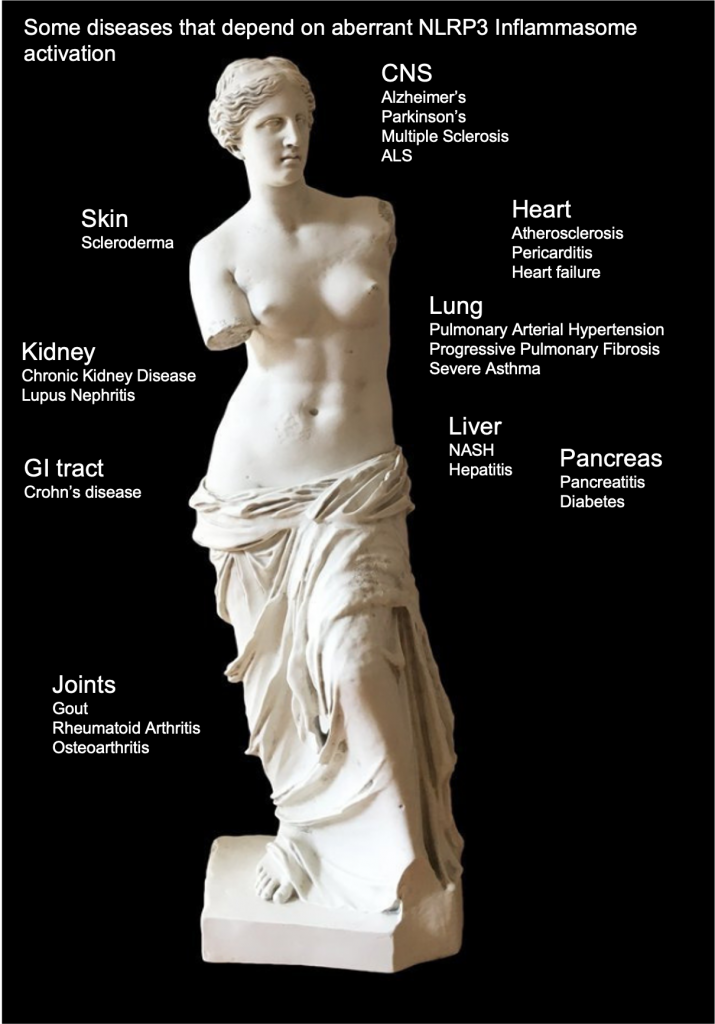
TARGETED DISEASES
Many chronic inflammatory and auto-immune diseases are being associated with continuously increased levels of MIF. These diseases tend to also depend on NLRP3 inflammasome activation.
Inhibition of MIF down-regulates multiple pro-inflammatory and fibrotic pathways and enhances sensitivity of the glucocorticoid receptor. Therefor Apaxen is focused on treatment of patients with Neutrophilic (non-allergic) Asthma, a disease that does not respond well to single cytokine treatment and is often resistant to corticosteroid-steroids.
Other pulmonary diseases that could benefit from our MIF inhibitors are COPD, IPF and Pulmonary Arterial Hypertension.
Results from these initial indications pave the way to many other indications and patient subsets.